Brunna Xavier Martins, Canada has been granted the IPTA Scientific Congress Award
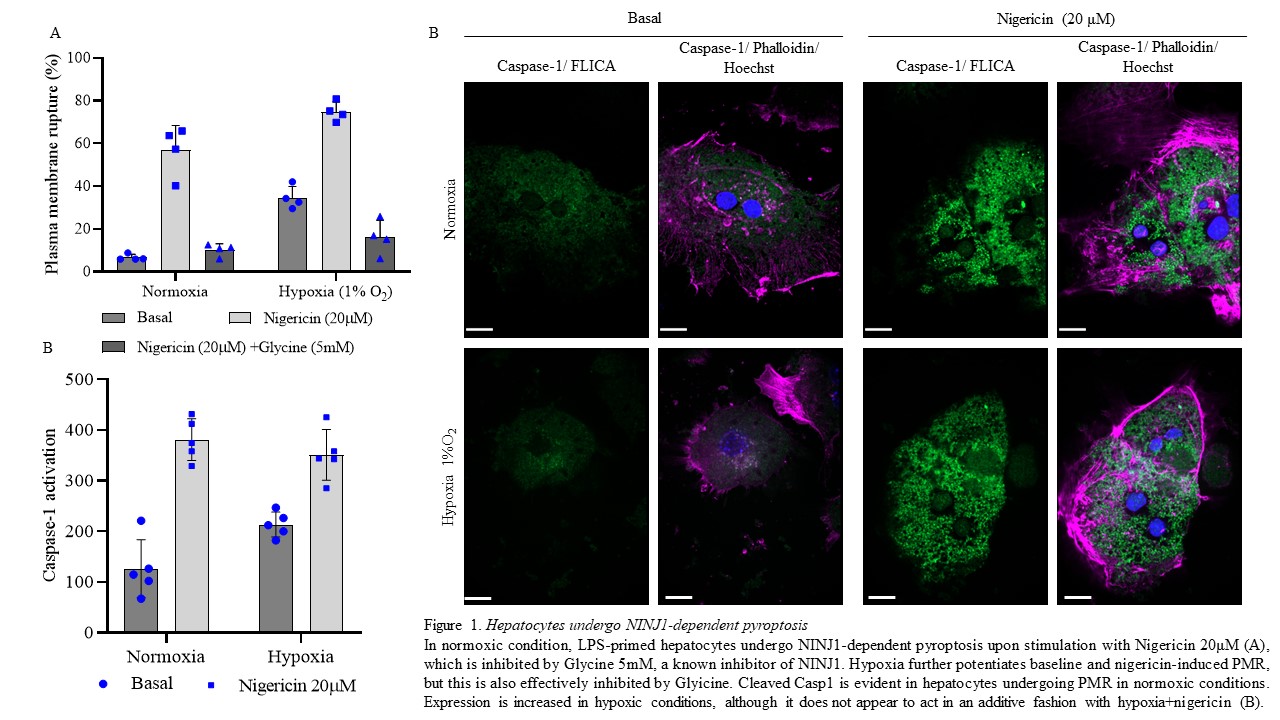
Ninjurin1 controls plasma membrane rupture in hepatocytes
Brunna Martins1, Jan Mossemann1, Danish Ali1, Sarah Zhao2, Gang Ye1, Philip J. Bilan1, Neil Goldenberg3, Benjamin Steinberg2,3, Blayne Amir Sayed1,4.
1Program in Cell Biology, The Hospital for Sick Children, Toronto, ON, Canada; 2Program in Neuroscience and Mental Health, The Hospital for Sick Children, Toronto, ON, Canada; 3Department of Anesthesia and Pain Medicine, The Hospital for Sick Children, Toronto, ON, Canada; 4Department of General and Thoracic Surgery, University of Toronto, Toronto, ON, Canada
Introduction: Hepatic ischemia-reperfusion injury (IRI) functionally limits the expansion of the donor pool, which negatively impacts both adult and pediatric candidates awaiting transplantation. Hypoxic stress during organ storage induces cellular injury and death, releasing damage associated molecular patterns (DAMPs) into the local environment during reperfusion. In the liver, DAMPs initiate pyroptosis, an inflammatory form of cell death, in which the NLRP3 inflammasome assembles and serves as a platform for caspase-1 (Casp1) activation. Subsequent Casp1-dependent cleavage of Gasdermin D (GSDMD) facilitates oligomerization of GSDMD N-terminal subunits into membrane pores, allowing release of pro-inflammatory cytokines (e.g. IL-1b), destabilization of the plasma membrane and eventual plasma membrane rupture (PMR). Kupffer cell pyroptosis contributes to hepatic IRI, but the role of hepatocytes is not clear. Recent data indicates that multiple forms of active PMR depend on Ninjurin1 (NINJ1), a transmembrane protein expressed in a variety of cell types, including hepatocytes. Here, we hypothesize that hepatocytes undergo NINJ1-dependent cell death during hypoxic injury and that inhibition of NINJ1 has the potential to directly limit hepatocyte injury.
Methods: An in vitro model of ischemia-reperfusion injury utilizing a hypoxic chamber (1% O2; Oxycycler TM, Biospherix) was used following isolation of primary murine hepatocytes (NINJ1+/+ or -/- mice, C57Bl/6 background). Classical pyroptosis was induced by lipopolysaccharide (LPS-1mg/ml) + Nigericin (20 μM) and PMR was measured by release of lactate dehydrogenase (LDH, Thermo Scientific), NINJ1 was inhibited by pretreatment with Glycine (5 mM). Casp1 cleavage was measured by confocal microscopy using the Caspase-1 substrate FLICA (Bio-Rad).
Results: Under normoxic conditions (21% O2), Hepatocytes undergo low levels of PMR, but induction of pyroptosis by LPS+Ng induces substantial increase (Fig 1A), which is strongly blunted by the addition of Glycine. Hypoxic injury significantly augments Hepatocyte PMR at baseline and with induction of pyroptosis, but NINJ1 inhibition effectively inhibits this response (Fig. 1A). This correlates with Hepatocyte expression of cleaved Casp1, which is induced under normoxic conditions by LPS+Ng, and also increased by hypoxic insult (Fig. 1B). Additionally, hypoxia increases mRNA expression of Ninj1, Gsdmd and Casp1 mRNA levels. Finally, Hepatocytes isolated from NINJ1-/- mice are protected from hypoxia- and pyroptotis-induced PMR.
Conclusions: Hepatocytes undergo PMR in an NINJ1-dependent fashion, and this response is augmented by hypoxic insult. NINJ1 inhibition by Glycine or genetic deletion protects Hepatocytes from lytic cell death induced by classical pyroptotic stimuli or hypoxia. These results identify a new potential target for ameliorating ischemia reperfusion injury during liver transplantation.